Goal of this exercise: Analyze the first study that used the CRISPR Cas9 system to successfully knock out an abnormal gene in human patients (Gillmore et al. 2021). Part 3 will examine potential off-target effects of the CRISPR-Cas9 system used to develop a treatment for ATTR amyloidosis.
Part 1: Overview of ATTR, CRISPR, and Gillmore et al. 2021
Part 2: Cut the TTR gene with Cas9 under the direction of guide RNA
Part 3: Look for potential off-target effects of Cas9
Part 4: Examine mutations of patients associated with the study
Part 5: Analyze frameshift mutations in monkeys and humans
Overview of Part 3
Off-target effects are defined here as changes in the genome caused by Cas9 cutting DNA at locations other than the target. Knockout studies attempt to design guide RNA that is highly specific to the gene of interest (the TTR gene in this case). In Part 3, the basic principle of an off-target effect will be illustrated via the search feature of Case It v7.0.4, using a 1M bp portion of chromosome 18 as the ‘subject’ DNA (the DNA that is searched). One potential off-target location that results from this search will be visualized using the NCBI Genome Data Viewer.
The entire human genome will then be searched for potential off-target locations via the BLAST procedure. One particular tool used to design guide RNA will be demonstrated, with results compared to the guide RNA used by Gillmore et al. 2021. Finally, the actual workflow used by Gillmore’s team to verify the specificity of their guide RNA will be shown.
Organization of Part 3
Steps 3-14: Look for potential off-target effects on a portion of chromosome 18.
Steps 15-21: Visualize location of one potential off-target effect using the NCBI Genome Data Viewer.
Steps 22-39 : Look for potential off-target effects by BLASTing the entire human genome.
Steps 40-45: Use Sythego Knockout Guide Design to compare efficiency of guide RNAs.
Step 46: Actual workflow used by Gillmore etal. 2012 to look for off-target effects.
1. If your browser is still open from Part 2, then minimize it or move it to the side so that you can work with Case it v7.0.4.
If you do not already have Case It v7.0.4, go to Step 1 of Part 2 for instructions on acquiring the software. If you are using Case It on a Mac, read these instructions for accessing files.
2. If Case It v7.0.4 is already open, quit the program and restart it. This is because you were working with a very large file in Part 2, so it is best to start afresh.
SEARCHING A PORTION OF CHROMOSOME 18 FOR OFF-TARGET EFFECTS
Goal of Steps 3-14: Use Case It v7.0.4 to look for potential off-target effects on a portion of chromosome 18 by searching for regions similar but not identical to the guide RNA spacer sequence. From Part 2, you know that there is only one location on chromosome 18 that perfectly matches the Cas9 target sequence used by Gillmore et al. 2021. Off-target sites can be sequences with less than 100% similarity, and we can find those sequences by setting our search parameters to be less than 100%.
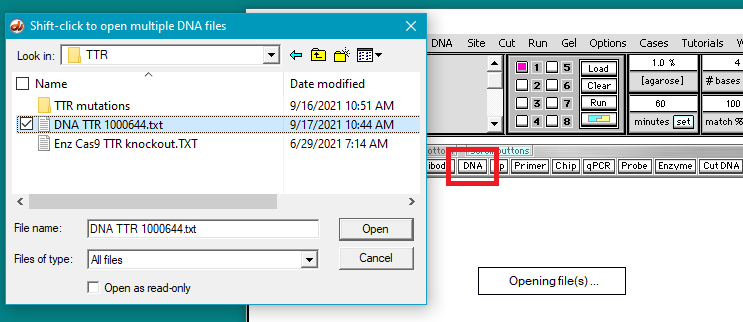
3. Use the DNA button to open the file DNA TTR 1000644.txt, located inside the TTR folder (Case It v704 PC > TTR folder).
Note: This file has consists of the 1,000,674 bases surrounding the TTR gene on chromosome 18. This file will be used instead of the 80 million base file used for Part 2, as the search feature of Case It v7.0.4 will not work with files over 2.67 million characters in size. Use of a file with one million characters will be suitable for our purposes here.
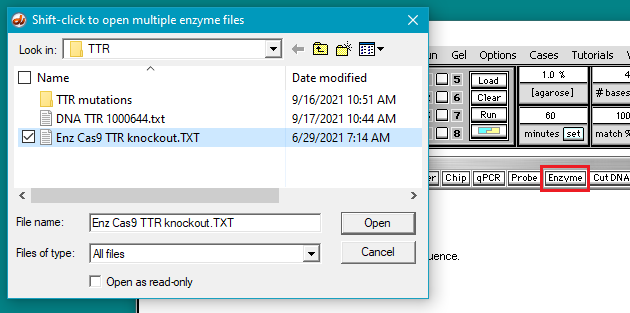
4. Use the Enzyme menu to open the file Enz Cas9 TTR knockout.TXT.
5. Double-click on the AAAGGCTGCTGATGACACCT sequence to highlight it, then right-click and select Copy > selected text to clipboard (only the letters will be copied).
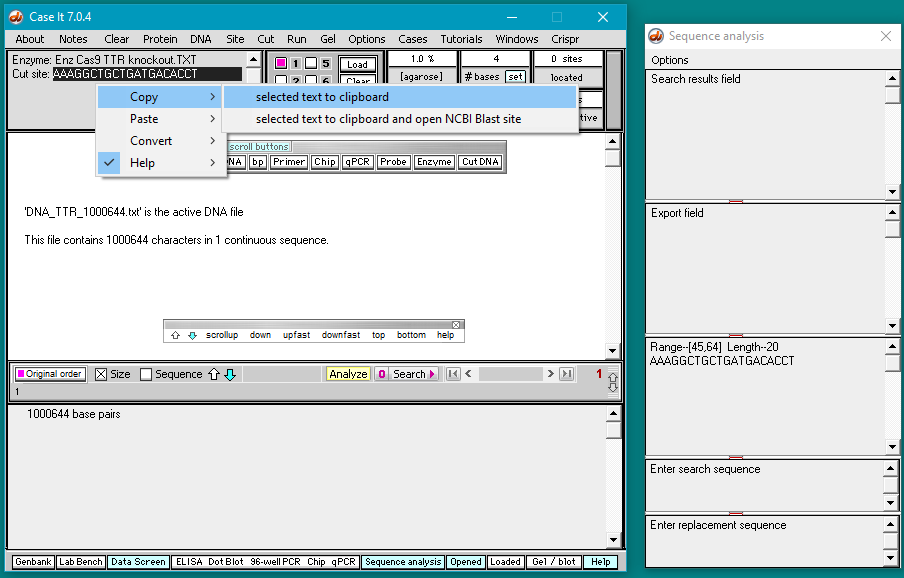
6. Right-click again at this same location and select Paste > clipboard into ‘search sequence’ field of Sequence Analysis window. This action will automatically paste the copied sequence into the Sequence Analysis window, second field from the bottom (in red box below).
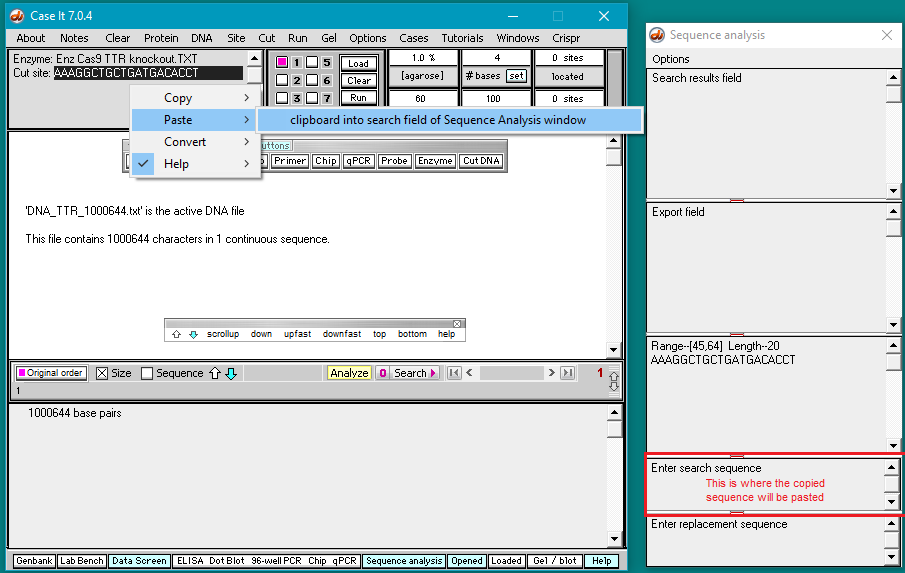
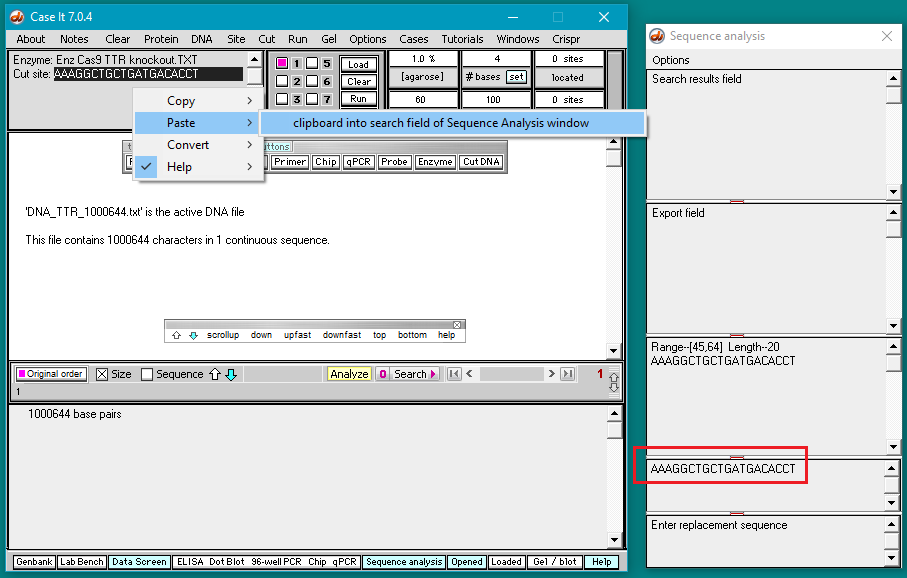
The sequence from the ‘enzyme’ field has been pasted into the ‘search sequence field’ (red box). Before the portion of chromosome 18 can be searched for this sequence, several additional steps are required (steps 7-9 below).
Goal of steps 7-14: To look for regions with at least a 70% match between a portion of chromosome 18 and the 20 base target sequence.
Rationale for choosing 70%: In reality, the last three bases (furthest from the PAM) can be mismatched without affecting Cas9 binding, and another three mismatches can be tolerated and Cas9 will still bind, as explained in this video. That works out to 70% (14 matches out of 20). Case It 7.0.4 will just look for 6 mismatches regardless of position, so the search routine is not entirely realistic but is suitable for our purposes here. Real-world applications require more sophisticated bioinformatics and experimental procedures to avoid off-target effects. For example, Gillmore et al. 2021 used a complex protocol to ensure that mismatches did not adversely affect patients in the study.
Note: The video linked above uses the term ‘protospacer’ to refer to the 20-base sequence on the non-target strand of DNA. The video should have referred to this as the ‘target site’ for Cas9, as the ‘protospacer’ is viral DNA that is the precursor to the ‘spacer’, stored in the bacterial immune system after the PAM sequence is removed – see Part 1.
7. Double-click in the match % field to highlight the number 100, then type in the number 70.
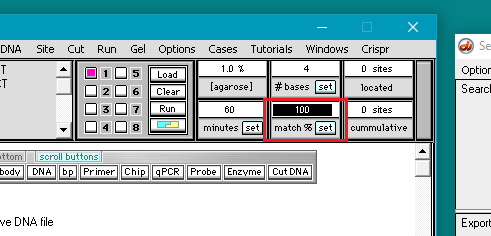
8. Click the set button and select Set match %.
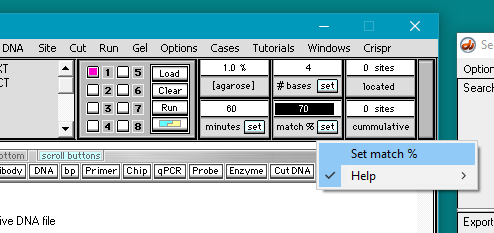
9. Before the DNA sequence can be searched by Case It v7.0.4, it must be loaded into a well*. To do this, click the Load button. The button next to the number 1 will turn blue, indicating that the well is now loaded, and the Loaded window will appear with a line indicating the contents of the well.
*Note: The Case It search routine will only work if DNA samples are loaded into wells first. This has nothing to do with running a gel, it is simply the way that the search routine works. Bioinformatics tools such as BLAST are used to search DNA sequence, but the Case It search routine has its advantages for this particular application, as will be seen.
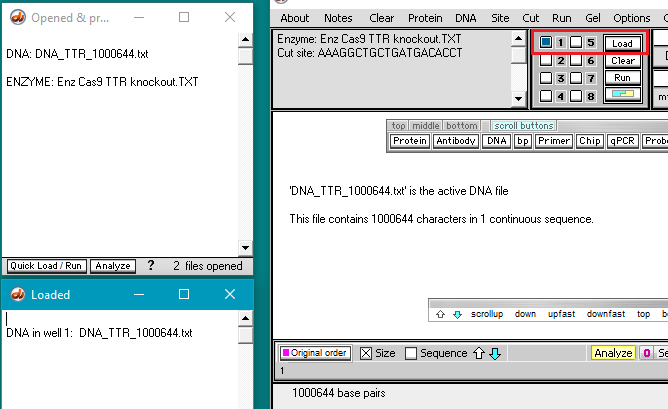
10. Click the Search button and select Search all loaded files, as shown below.
Important! Wait until the Searching sequence(s)… box disappears before doing anything else (it will take about a minute to complete the search). As this happens, each hit on the search sequence will be briefly highlighted.
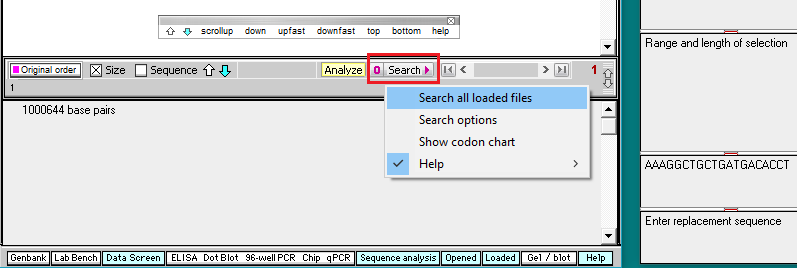
11. The DNA sequence has been searched for all matches that have at least 70% of the bases matching the search sequence. Percent matches and locations are shown both in the Sequence Analysis window and also in the upper field on the main screen.
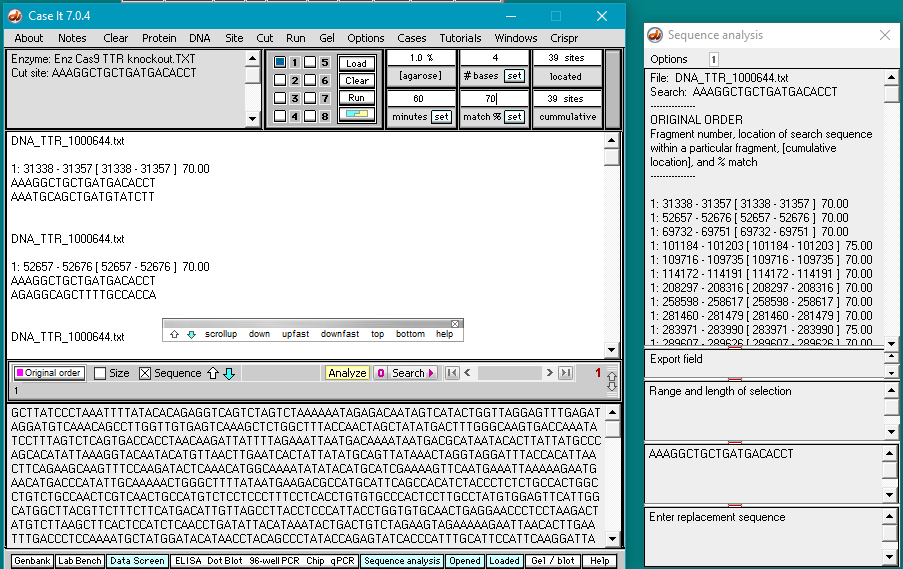
12. Clicking on a line in the Sequence Analysis (SA) window locates and highlights that sequence in the lower field of the main screen. Scroll down on the sequence analysis window and click on the line showing a 100% match. That sequence will be highlighted in the lower screen of the main window. Note that there is a PAM sequence (GGG) next to it. Recall that Cas9 requires an 5′-NGG-3′ sequence be next to the target site, where N can be any of the four bases (A,T,C, or G). Sequences are always displayed in the 5′ to 3′ direction, so the PAM (if present) must be to the right (3′ side) of the highlighted sequence.
Also scroll down (or use the dragbar) on the large field of the main window to find the corresponding entry for the 100% match. This will be highlighted in black.
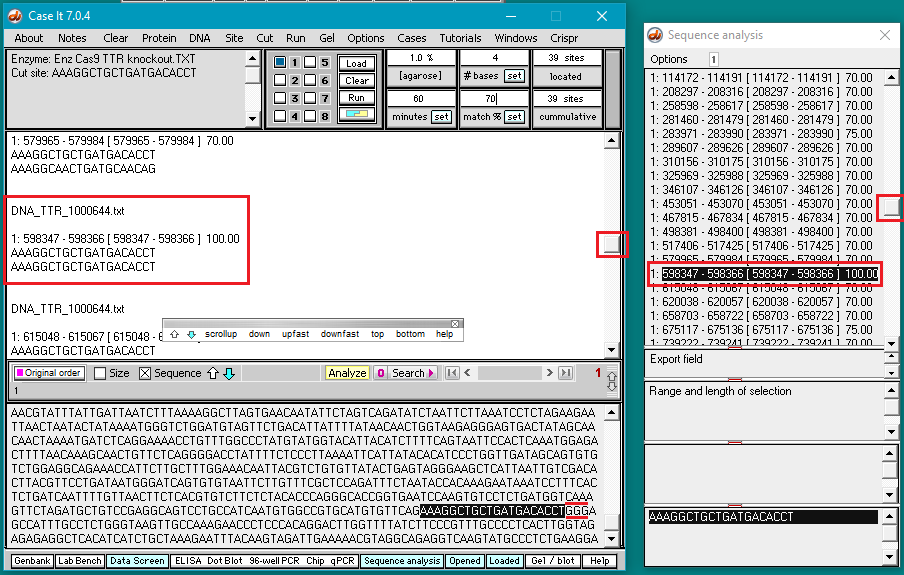
13. Scroll back to the top of the search results in Sequence Analysis window and click on each line to see if a NGG (PAM) sequence is present next to the selected sequence. Note that the 11th line down (showing the sequence at position 289607) has a 70% match, and is followed by GGG (see red box at bottom of image). Scroll in the large field on the main screen to find the corresponding entry (at position 289607).
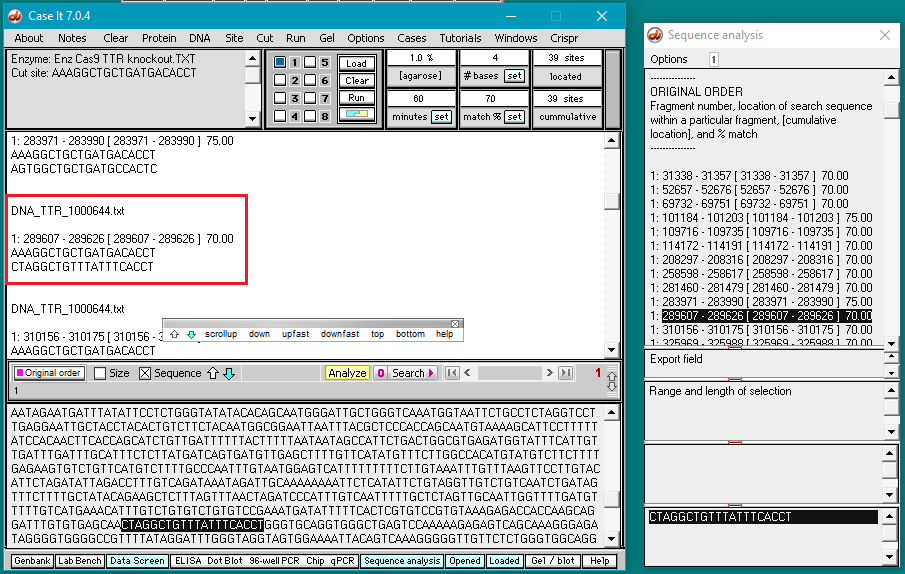
Question: There are four mismatches within 12 bases of the PAM. Look again at information on guide RNA design reproduced below from the AddGene CRISPR Guide, and compare the mismatch above with the 100% match below. Based on this information, how likely is it that Cas9 would make an off-target cut at this particular location?

“Cas9 will only cleave a given locus if the gRNA spacer sequence shares sufficient homology with the target DNA. Once the Cas9-gRNA complex binds a putative DNA target, the seed sequence (8-10 bases at the 3′ end of the gRNA targeting sequence will begin to anneal to the target DNA. If the seed and target DNA sequences match, the gRNA [spacer sequence] will continue to anneal to the target DNA in a 3′ to 5′ direction [right to left in the above diagram]. Thus, mismatches between the target sequence in the 3′ seed sequence completely abolish target cleavage, whereas mismatches toward the 5′ end distal to the PAM often still permit target cleavage.” Source: AddGene CRISPR Guide
Note: The 3′ to 5′ direction in the above quote refers to the ends of the guide RNA spacer sequence and the ‘non-target strand’ of DNA, which are identical except for the U’s in the spacer sequence being replaced by T’s in the DNA sequence.
14. The 70% match site above was found on a sequence representing a portion of chromosome 18. This corresponds to position 31284248 on the complete sequence of chromosome 18*. This number will be used in Step 15 below.
*Calculation: On the portion of chromosome 18 that was searched, the 70% match site began at position 289607 and the 100% match site began at 598347, for a relative difference of 308740. The location of the 100% match site on the complete sequence for chromosome 18 is 31592988 (from Part 2). Therefore, the location of the 70% match site on the complete chromosome 18 sequence is 31592988 – 308740 = 31284248.
VISUALIZE LOCATION OF ONE POTENTIAL OFF-TARGET EFFECT
Goal of Steps 15-21: To visualize the location of one potential off-target effect using the NCBI Genome Data Viewer, and to determine if this location falls in an exon (expressed region of a gene) or an intron (regions between exons). Off-target effects in exons are potentially more disruptive than effects in introns, because mutations in exons directly affect protein structure. Off-target effects in introns or intergenic regions cannot be ignored, however, because introns are involved in gene expression, and intergenic regions (regions between genes) may have similar functions. In reality, complex experimental and computational methods are required to ensure that a particular guide RNA will knock out an abnormal gene without causing other adverse effects in the genome.
15. Reopen the Genome Data Viewer that was used in Part 2. If you have closed it, right-click on this link and open it in a new tab or window. Then click the Browse genome button.
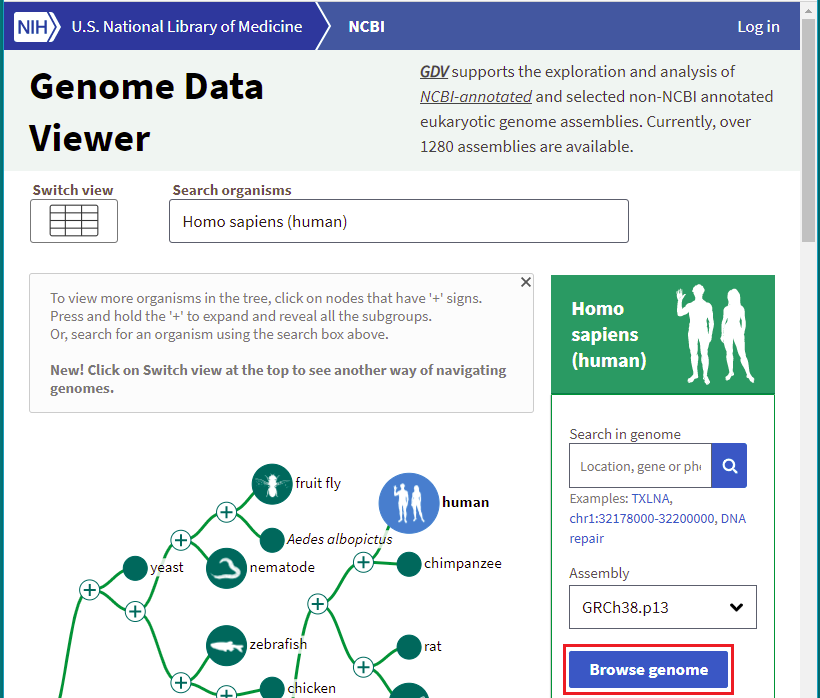
16. Type 18:31284248 into the ‘Search assembly’ field. This is the beginning of the 70% match site from Steps 13-14 above (CTAGGCTGTTTATTTCACCT followed by a GGG PAM site, circled in red).
17. The Genome Data Viewer (GDV) also enables you to search by sequence rather than location. To do this, click the Tools menu and select the Search option.
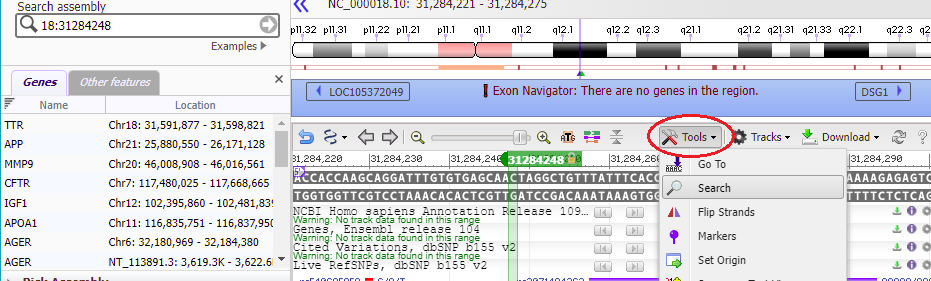
18. Copy CTAGGCTGTTTATTTCACCT to your clipboard, right-click to paste the sequence into the search box, and click OK.
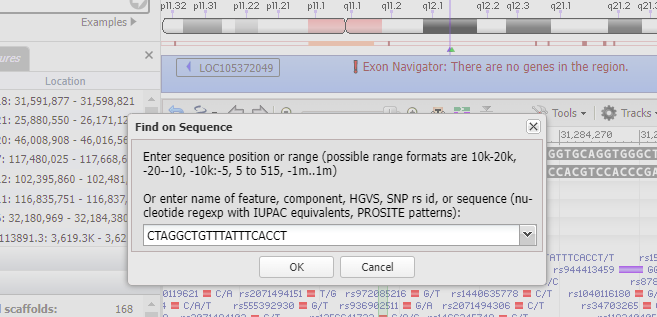
19. Although it says there are no search results to display, if a green check mark appears on the ‘Sequence tab’ then that means the sequence was found on chromosome 18 (first image below). Click on the green check mark, and the found sequence will appear with its location given (second image below).
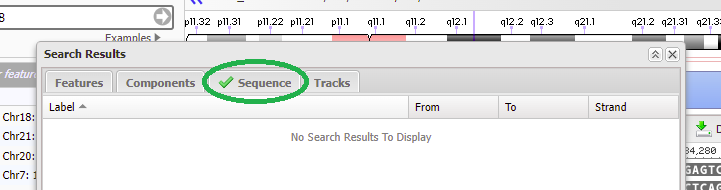
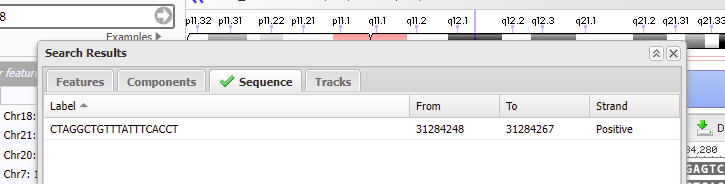
20. Double-click on the sequence, and the found location will be shaded green. Click Close to close the box.
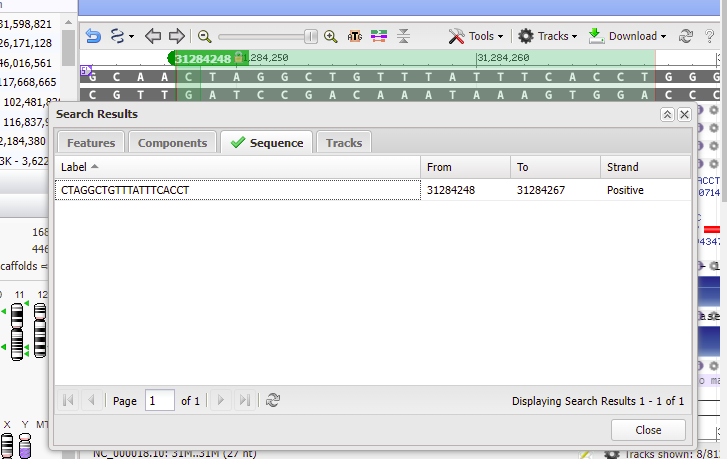
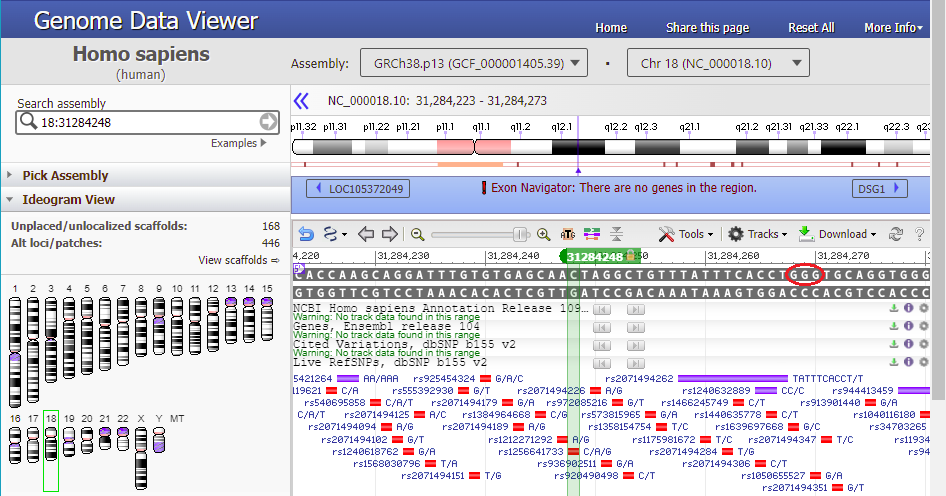
21. Text on the purple bar indicates that there are “no genes in this region”.
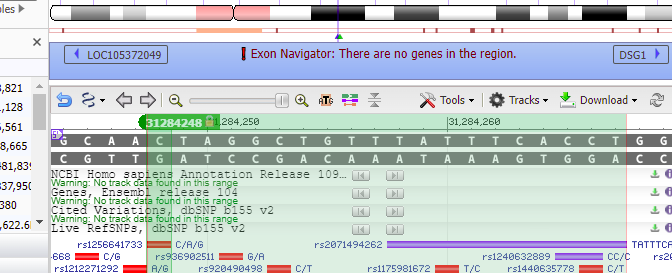
Use the slider to slowly zoom out, and note that the found location is not associated with any gene (green lines with arrows). Keep in mind that this was just one potential off-target location, used as an example. The next series of steps will make a much more extensive search of the entire human genome.
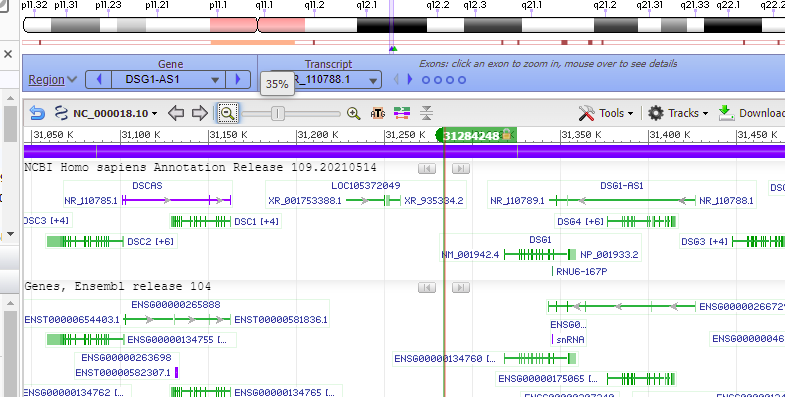
Question: Note that the off-target effect is not found directly in a gene (designated here by colored lines). Does this mean that an off-target cut here will have no harmful effect?
SEARCHING ENTIRE HUMAN GENOME FOR OFF-TARGET EFFECTS
Goal of Steps 22-39: Look for potential off-target effects by BLASTing the entire human genome with the Cas9 target sequence used by Gillmore et al. 2021. Recall that in Part 2, there was only one exact match for this 20-base sequence in chromosome 18. The procedure below will look for exact and partial matches for the target sequence in all chromosomes of the human genome.
22. Copy AAAGGCTGCTGATGACACCT and right-click here to go to the NCBI BLAST site (select ‘open in new tab’ or ‘open in new window’).
23. Paste the sequence into the query sequence field as shown below. Click the circle next to Genomic + transcript databases, which will automatically select Human genomic plus transcript (Human G_T) as the database to be searched. Then click the BLAST button.
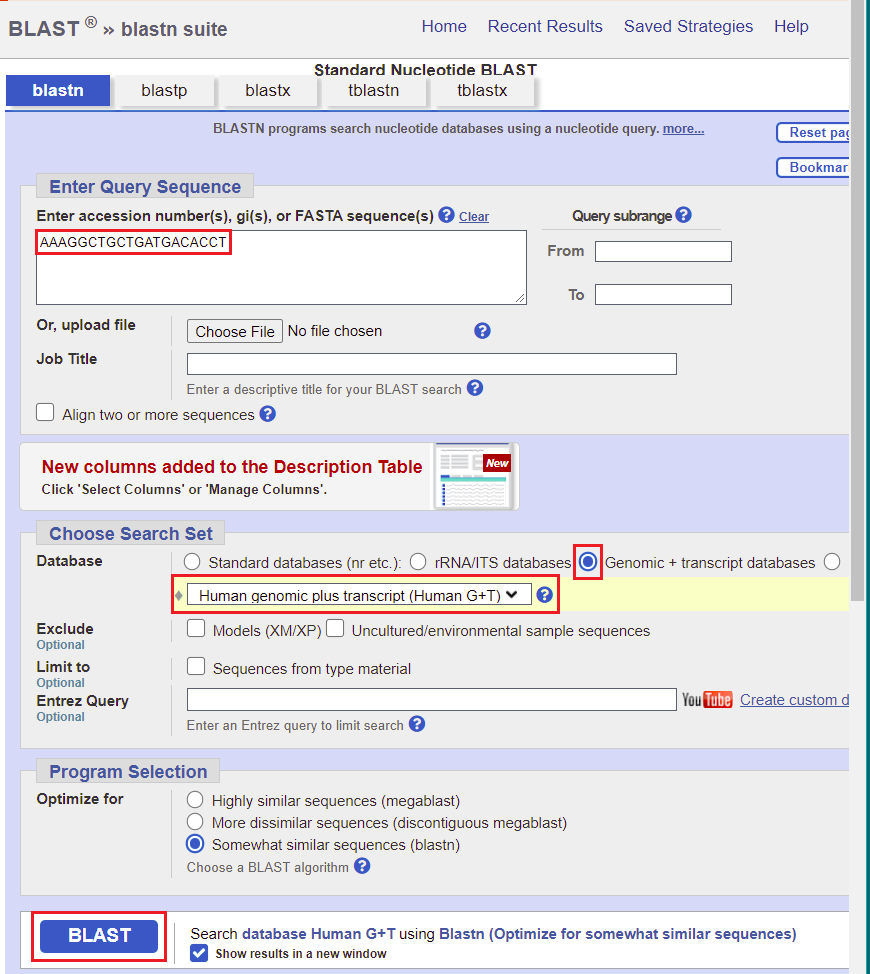
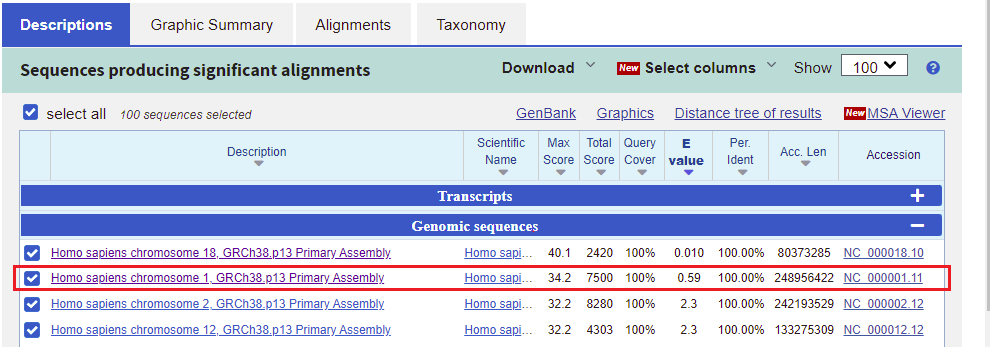
24. A page will appear giving BLAST results, with the lowest E-values and the highest scores listed first*. The best match under Transcripts is for Homo sapiens transthyretin (TTR) mRNA. So that is promising, but what we are really interested in are searches involving entire chromosomes. To hide the Transcript results and see results for Genomic Sequences, click the minus sign on the right-hand side of the blue bar.
*Note: When interpreting BLAST results you want to look for relatively low E-values and relatively high scores. The E-value is a measure of how many matches could be found by chance alone, and the score is a measure of how large a database has to be for a particular match to be found by chance alone.
25. Again, the top entry has the lowest E value and highest score. Click on the first link, for chromosome 18.
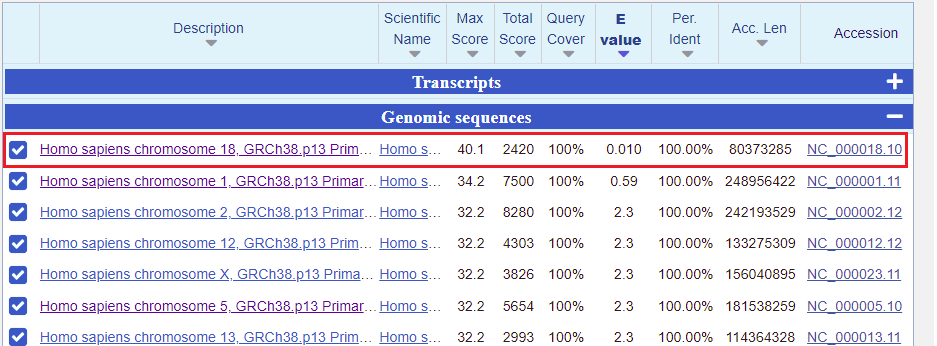
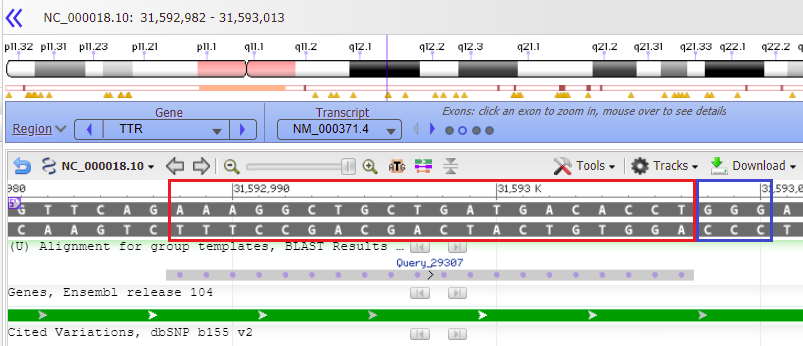
26. All of the matches for chromosome 18 are listed, with the best match first. Note that this is the only one that perfectly matches all 20 bases of the search query. It has low expected value, meaning that it is unlikely this result occurred by chance, and a relatively high score (bit score), which is a measure of how large a data set would have to be to get a result like this by chance. All 20 bases of the Query match the 20 bases on chromosome 18 (the Subject, abbreviated Sbjct), so the Identities value is 20/20 (100%). There are no gaps in the match, meaning no missing bases.
Note: The Strand is Plus/Plus, meaning that that the numerical ordering of the query and subject both increase in value going from left to right (1 to 20 for the query, 31592988 to 31593007 for the subject). This means that the query sequence was found on the subject sequence (the sequence originally entered into Genbank) rather than on the sequence that would have been complementary to it. BLAST searches both the original strand and its complementary strand, in the 5′ to 3′ direction.
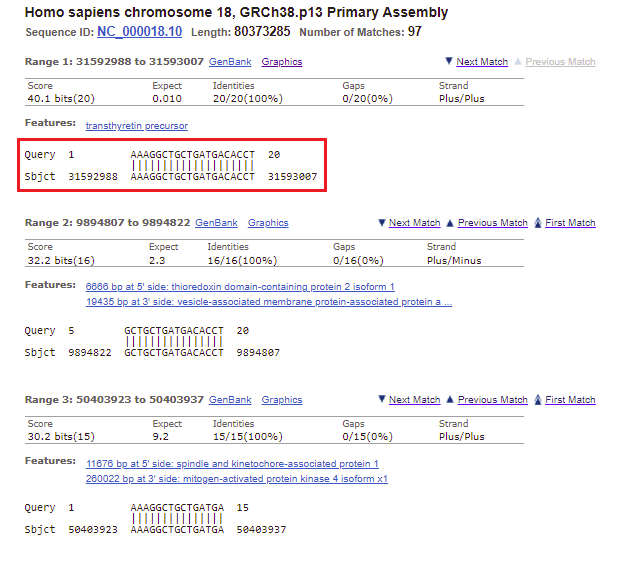
27. Select the range 31592988 to 31593007 as shown below, then right-click and copy the range to your clipboard.
Locate the Genome Data Viewer link on the right-hand side of the page, then right-click on it and select either ‘open link in new tab’ or ‘open link in new window’
28. Make sure that the chromosome 18 icon is selected in the ‘ideogram view’ on the left side of the screen, then click the Tools button and select Go To.
29. Paste what you copied in Step 27 into the box, and click OK.
30. Results are shown below with a GGG sequence to the right (in blue box). Compare this result with that shown in Step 44 of Part 2 and also Steps 16-20 of Part 2.
31. Go back to the BLAST results tab of your browser, scroll up the page, and click on the Descriptions tab. Note that the second entry on the list has an E-value less than one (0.59), on chromosome 1. Click on this line, as shown below.
32. The best match for chromosome 1 matches the first 17 bases of the search query, but does not match the last three bases.
Note: The Strand is Plus/Minus, meaning that that the numerical ordering of the subject goes in reverse order (10962005 to 1096189). This means that the query was found on the sequence complementary to the subject sequence.
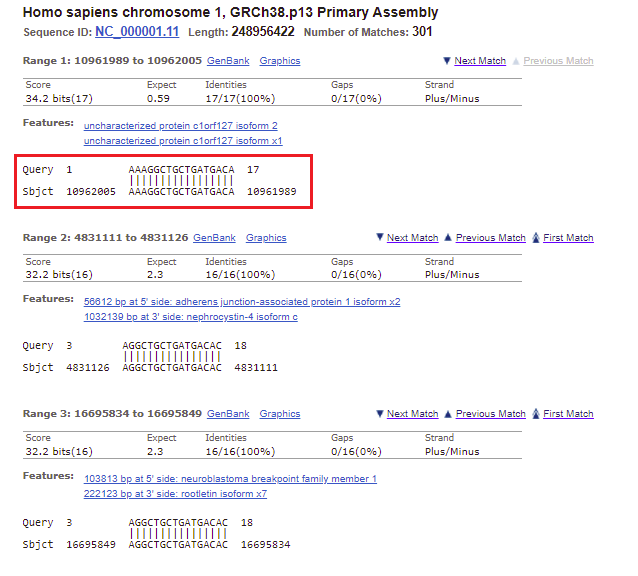
33. Go back to the Genome Data Viewer tab of your browser and click on the icon for chromosome 1 (far left), and then click the Tools button and select Go To. Chromosome 1 is three times larger than chromosome 18, with 249 million bp.
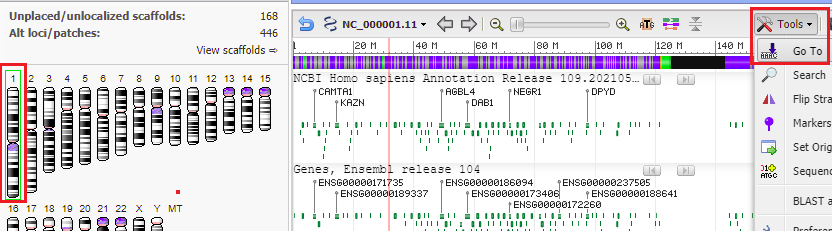
34. The location of the top match for chromosome 1 was 10961989 to 10962005 (detail from image in Step 32 reproduced below).
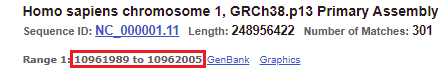
35. Copy and paste this range into the Go To box, and click OK.
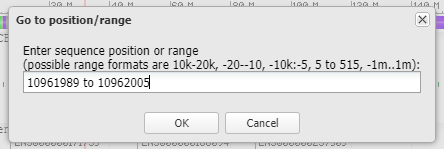
36. Results are shown below, with the located sequence on the complementary strand of the original subject sequence (in red box).
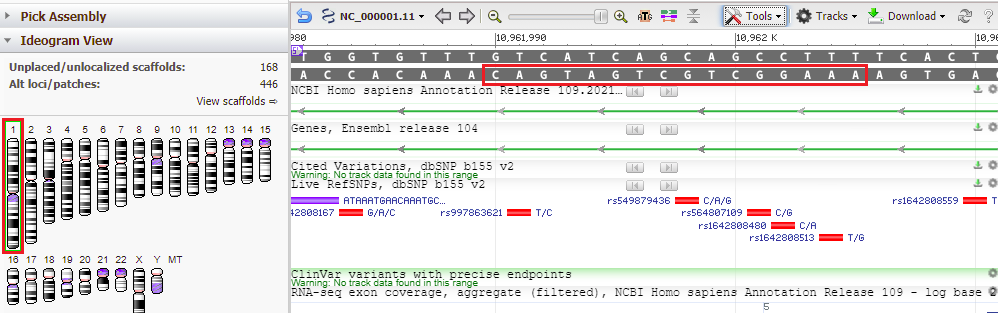
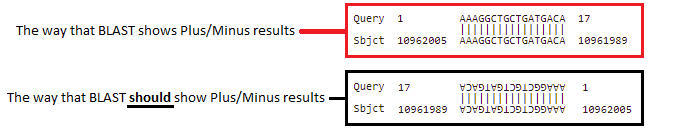
Note: The search query was AAAGGCTGCTGATGACA, and this was found on the complementary strand, so it is in reverse order – CAGTAGTCGTCGGAAA). This is because BLAST searches both DNA strands, the one originally entered into Genbank and its complement, in the 5′ to 3′ direction (5′ is shown on the image above, in purple).
The way BLAST shows a Plus/Minus result is confusing. The way BLAST shows the result is in the red box below, but the way BLAST should show the result is upside-down and backwards, as in the second image below. This would clearly indicate that the query was found on the strand complementary to the one entered into Genbank.
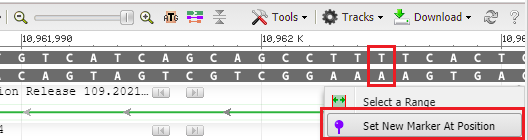
37. To see where this particular location falls in the gene sequence, first click at the location of the red box below, then right-click and select Set New Marker At Position. Click OK in the box, and a marker will appear at location 10962005.
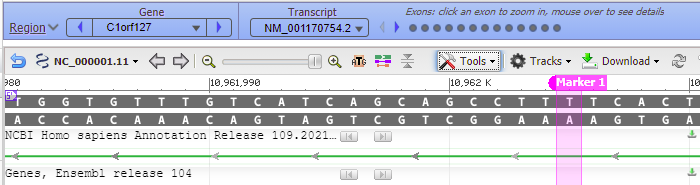
38. Move the slider to the 60% position…
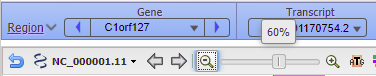
39. …and the location of the marker will be visible, showing that the location is in an intron of a gene (narrow green line) whereas the exons are shown by the green rectangles.
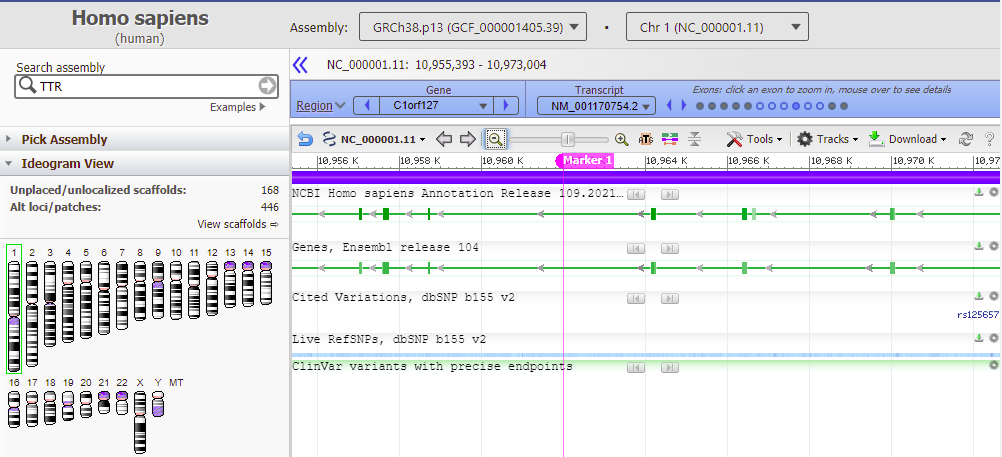
Question: Will Cas9 cut chromosome 1 at this location? What are the factors that determine whether Cas9 will cut DNA at a particular location? If it did cut here, would it necessarily ‘knock out’ this gene? Note that the gene is named C1orf127. Here are links to the gene in Wikipedia and also in the NCBI Gene database. What would be the effect of knocking out this particular gene?
USING SYTHTHEGO KNOCKOUT GUIDE DESIGN TOOL
Goal of Step 40-45: To use a knockout design tool developed by Synthego, a biotechnology company based in California. To determine if this tool identifies the same guide RNA that Gillmore’s group used to knock out the TTR gene.
40. Use the CRISPR menu to open Synthego knockout guide design, or right-click here and select ‘open in new tab’ or ‘open in new window’.
41. Select Homo sapiens, type TTR in the Gene field, and click Search.
42. Click the All Guides tab.
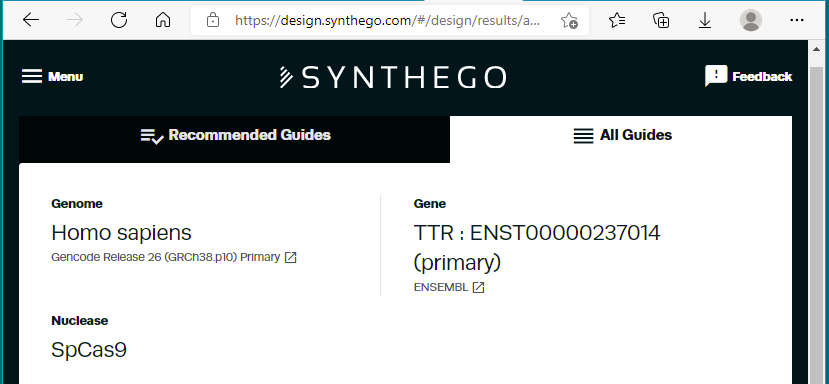
43. Scroll down tothe 13th-ranked guide RNA sequence identified by their design tool. Click the guide sequence to ‘validate this guide’.
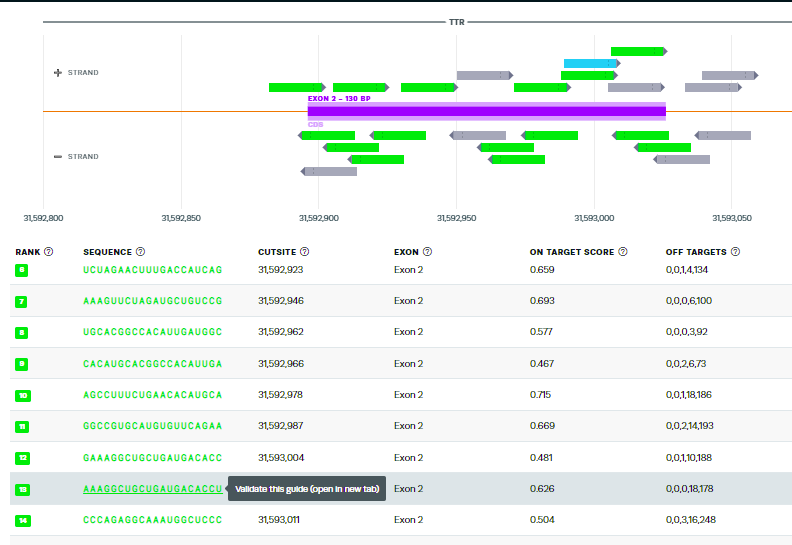
44. This is the same guide RNA used by Gillmore’s team, but ranked 13th of the possible guides by the Synthego design tool. Note that the tool identifies sites on other chromosomes with potential mismatches, and gives the genes (if present) for those mismatched regions, along with the specific mismatches in each sequence. Tools at the NCBI site can be used to obtain more information about the function of these genes.
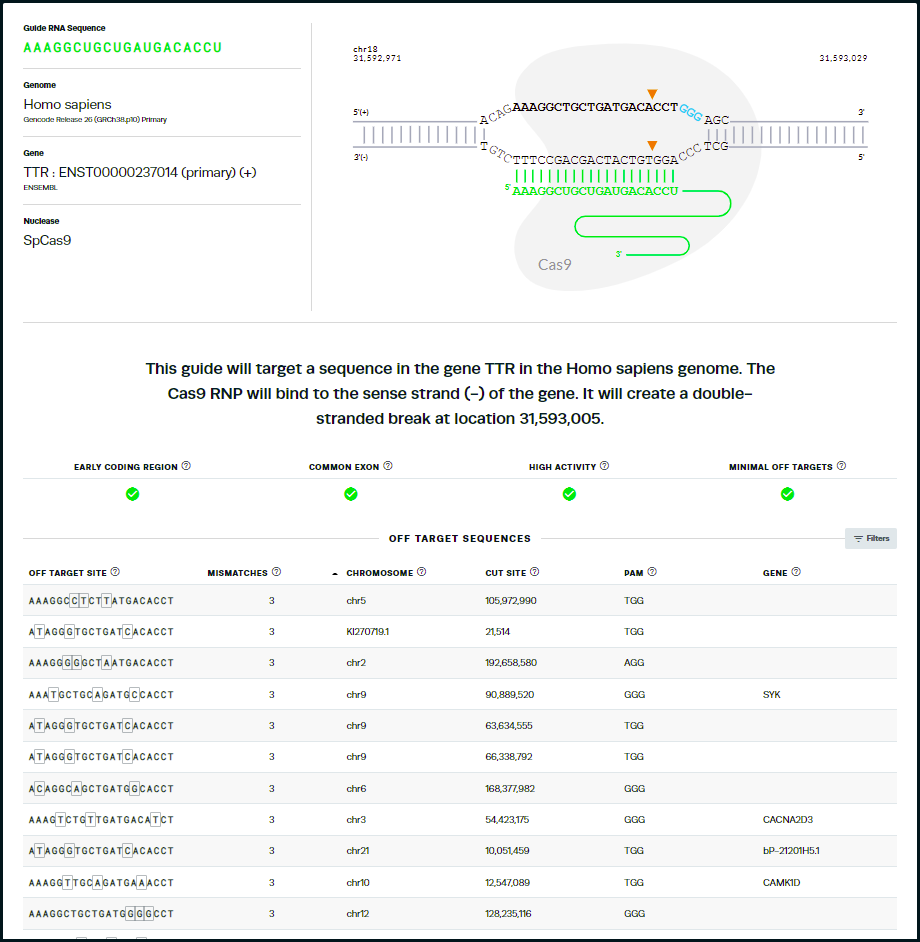
Questions: Does this result necessarily mean that Gillmore’s team used the ‘wrong’ guide RNA to knock out the TTR gene? What are some of the reasons why Gillmore’s team used a guide RNA sequence different from the ones that Synthego’s tool recommended?
45. This is but one of many tools to find ‘optimal’ guide RNA sequences for gene editing that minimize off-target effects of Cas9. There is a very large literature on this and other aspects of CRISPR technology, which you are encouraged to explore.
Goal of Step 46: To see summary of actual procedure used by Gillmore’s team to examine potential off-target effects.
46. Gillmore et al. 2021 used computational methods and a series of in vivo and in vitro experiments to make sure that the guide RNA sequence they used would not cause harmful off-target effects and be safe for use in human patients.
Summary of key points in linked document
Page 4. Used lipid nanoparticles to deliver guide RNA and Cas9 messenger RNA to liver cells (Cas9 was transcribed by the cell from mRNA).
Page 7. Procedure used to determine optimal dose via phase 1 study in humans.
Page 8. Nonclinical studies of effect on human cells in vitro, and non-human cells in vivo (mice and non-human primates).
Page 9. Photos showing reduction in TTR protein in transgenic mice (mice with the TTR mutation).
Page 10. Graph showing reduction in TTR protein in non-human primates.
Pages 13-14. Safety and toxicity evaluation.
Page 17-18. Discovery and validation of potential off-target edits via computation prediction, SITE-seq and GUIDE-seq and next generation sequencing (NGS).
P. 21. Of 658 potenial off-target sites, 7 were validated, none of which occurred in exons (expressed regions of genes).
P. 22. No detectable off-target editing at concentrations up to 3X the pharmacologic dose.
Top of this page
Go to Part 1: Overview of ATTR, CRISPR, and Gillmore et al. 2021
Go to Part 2: Cut the TTR gene with Cas9 under the direction of guide RNA
Go to Part 4: Examine mutations of patients associated with the study
Go to Part 5: Analyze frameshift mutations in monkeys and humans